软件功能
1.数据库管理
选择数据库|经理命令打开数据库管理器”对话框
2.DNA和蛋白质数据库
dnaman 9该软件的数据库功能,允许用户组织DNA和蛋白质序列的不同科目
3.编辑记录信息
有关特定记录的信息可以编辑。在序列列表框中,所有记录按字母顺序或记录顺序列出。每个记录名字的数量显示记录的记录号ID.用DNAMAN自动分配。因此,您可以为不同的记录使用相同的名称
4.扫描序列的相似性
它扫描所有的序列记录在默认的数据库搜索当前序列的同源序列。如果默认数据库包含DNA序列,则默认序列必须是DNA序列。如果默认数据库包含蛋白质序列,则默认序列必须是蛋白质序列。DNAMAN将使用快速对准方法扫描数据库的序列相似性。您可以选择对结果的最终输出使用快速对齐或最佳对齐方式
5.错配分析
错配分析的目的是找到所有可能的退火位点的DNA序列的引物在默认。此功能可用于PCR和DNA测序的引物选择。权重矩阵用来区分引物位置的重要性。由于引物在3’末端对目标DNA的匹配比PCR扩增的5末端更重要,所以更多的权重被赋予3’末端。为了提高PCR引物的特异性,应始终检查引物与靶DNA之间是否存在次级退火位点
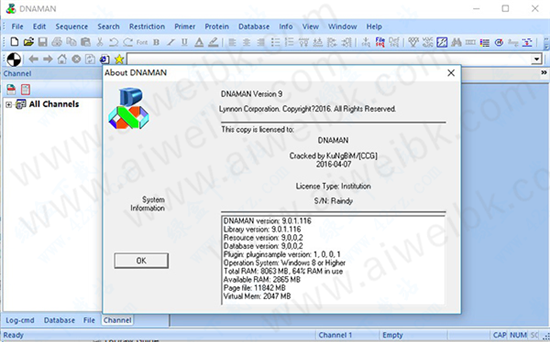
dnaman 9怎么用
1.安装完成后将破解补丁复制到安装目录下替换源文件即可,一般默认安装目录为:C:\Program Files (x86)\DNAMAN
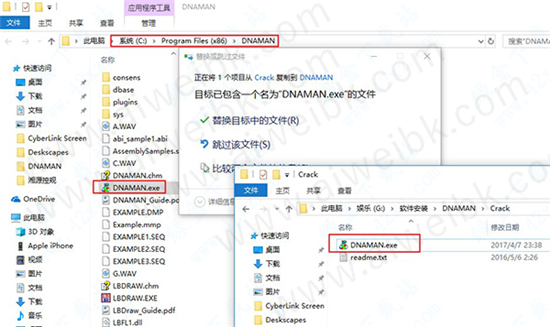
2.破解完成
软件特色
1.DNA和蛋白质序列编辑
2.DNA序列转化
3.多序列比对,对齐编辑和分析
4.系统树分析
5.DNA或蛋白质序列的点阵比较
6.DNA序列组装和编辑
7.BLAST通过网络界面在Intranet / Internet Server上进行搜索
8.序列和数据库中的增强型图案搜索
9.SiRNA选择器
10.限制分析
11.绘制序列图与出版品质
12.限制模式预测
13.电子克隆
14.从限制片段重建限制图
15.静音突变分析创建/破坏限制性位点
16.导向不匹配以创建/破坏限制站点
17.翻译和密码子使用分析
18.蛋白质疏水性/亲水性分析
19.蛋白质表征:等电点的序列组成和预测
20.蛋白质二级结构预测
21.反向翻译
22.PCR和测序引物的设计
23.表征DNA或引物序列的热力学性质
24.分歧分析
25.管理寡核苷酸,DNA和蛋白质数据库
26.生成随机序列
dnaman 9使用教程
一、将待分析序列装入Channel
1.通过File|Open 命令打开待分析序列文件,则打开的序列自动装入默认Channel。(初始为 channel1)可以通过激活不同的channel(例如:channel5)来改变序列装入的Channel2.通过Sequence|Load Sequence 菜单的子菜单打开文件或将选定的部分序列装入Channel。
dnaman 9可以通过Sequence|Current Sequence|Analysis Defination 命令打开一个对话框,通过此对话框可以设定序列的性质(DNA 或蛋白质),名称,要分析的片段等参数
二、以不同形式显示序列
1.通过Sequence|Display Sequence 命令打开对话框
2.根据不同的需要,可以选择显示不同的序列转换形式,可选择:
1)显示序列和成分
2)显示待分析序列的反向序列
3)显示待分析序列的反向互补序列
4)显示待分析序列的互补序列
5)显示待分析序列的双链序列
6)显示待分析序列的对应RNA序列
3.参数说明如下
Results 分析结果显示
其中包括:Show summary(显示概要) Show sites on sequence(在结果中显示酶切位点)
Draw restriction map(显示限制性酶切图)Draw restriction pattern(显示限制性酶切模式图)
Ignore enzymes with more than(忽略大于某设定值的酶切位点)
Ignore enzymes with less than(忽略小于某设定值的酶切位点)
Target DNA (目标DNA 特性)
circular(环型DNA),dam/dcm methylation(dam/dcm 甲基化)all DNA in Sequence Channel(选择此项,在Sequence Channel 中的所有序列将被分析, 如果选择了Draw restriction pattern,那么当所有的channel 中共有两条DNA 时,则只能选择两个酶分析,如果共有三个以上DNA 时,则只能用一个酶分析。
三、dnaman 9序列同源性分析
1)两序列同源性分析
1.通过Sequence|Two Sequence Alignment 命令打开对话框
2.参数说明如下
Alignment method 比对方法,通常可选Quick(快速比对)或Smith&Waterman(最佳比对),当选择快速比对时,设置较小的k-tuple 值,可以提高精确度,当序列较长时,一般要设置较大的k-tuple 值。(dna 序列:k-tuple 值可选范围2—6;蛋白质序列:k-tuple 值可选范围1—3
2)多序列同源性分析
1.通过打开Sequence|Multiple Sequence Alignment 命令打开对话框
2.参数说明如下
a.从文件中选择参加比对的序列
b.从文件夹中选择参加比对的序列
c.从channel 中选择参加比对的序列
d.从数据库中选择参加比对的序列
e.清除选择的序列(鼠标点击左边显示框中的序列名选择)
f.清除全部序列